









研究者総覧「情報知」
複雑系科学専攻
- 氏 名
- 北浦 和夫(きたうら かずお)
- 講座等
- 大規模分子計算論
- 職 名
- 客員教授
- 学 位
- 理学博士
- 研究分野
- 量子化学

研究内容
大規模分子系の電子状態計算法の開発
Ab initio MO法や密度汎関数理論などの電子状態計算法は、分子・分子集合系の構造・性質・反応の研究で広く活用されている。コンピューターの性能向上と理論・アルゴリズムの発展に伴い、適用対象が継続的に拡大してきた。近年、京コンピュータのような超並列計算機が利用可能になったことで、従来困難であった超高精度計算や大規模系計算などが可能になり、電子状態計算による研究の新たなステージが到来しつつある。計算シミュレーションの永遠の課題は計算結果の信頼性の向上であるが、このためには高精度計算理論とあわせて現実系に近いモデルを計算対象にする必要がある。後者を追求すると、しばしば、巨大系になる。巨大系の電子状態計算法の研究は、1990年はじめから活発に行われるようになり、私たちも、タンパク質など数千から数万原子からなる巨大分子・分子系の電子状態計算を可能とするフラグメント分子軌道法(FMO法)の開発を行っている。
FMO法は、模式図に示すように、巨大分子系を小さなフラグメントに分割し、フラグメントとフラグメントペアについてab initio MO計算を行い、それらの計算結果を用いて全系のプロパティを求める方法である。
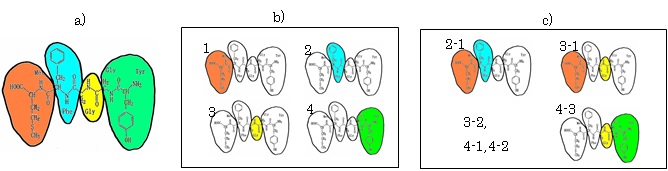
FMO計算法の模式図。a)ポリペプチドを、b)フラグメントに分割し、これらについて電子状態計算を行う。さらに、c)フラグメントペアについても電子状態計算を行い、両者の結果を用いて全系a)のプロパティを計算する。
FMO法の開発は、Hartree-Fock法から始めて順次各種電子相関理論計算をサポートし、現時点では、小型・中型分子の計算でよく用いられている電子状態理論をほぼカバーしている。また、解析的gradientによる構造最適化、解析的hessianによる基準振動解析と遷移状態計算、連続誘電体溶媒モデルによる溶媒和自由エネルギー計算、FMOと古典力場の融合計算による構造最適化も可能である。これらのFMO計算法は、GAMESS(http://www.msg. ameslab.gov/GAMESS/)に組み込んで公開している。
FMO法はタンパク質や分子クラスターなどの計算に応用されている。以下にタンパク質の計算の一例として、インフルエンザ感染の初期過程に関連する、ウイルスの表面タンパク質と宿主細胞表面のシアル酸の結合自由エネルギー計算について紹介する。計算は、ヘマグルチニンとシアル酸複合体の結晶構造データ(PDBID:1HGG、3量体)からモノマー(原子数は約8,260)のデータを取り出し、MDシミュレーション(AMBER99/GLYCAM6e/TIP3P)で構造モデルを作成し、FMO-PCM/6-31G(d)レベルで結合自由エネルギーを計算した(PCM:polarizable continuum model)。計算は、トリ型ヘマグルチニン、ヒト型ヘマグルチニンとトリ型シアル酸、ヒト型シアル酸の組み合わせについて行い、トリ型はトリ型、ヒト型はヒト型どうしで結合エネルギーが大きく、宿主域変異と矛盾しない結果が得られた。
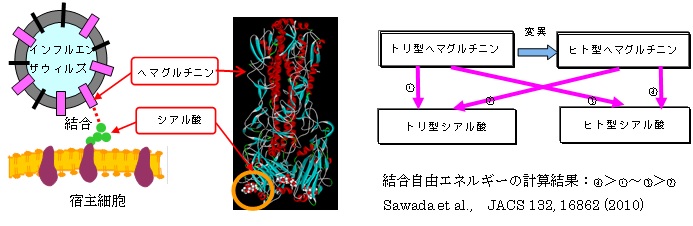
a)ヘマグルチニンとシアル酸の複合体構造(PDB ID:1HGG)とFMO-PCM計算から求めた結合自由エネルギーの大きさの順序。
上記計算はPCクラスターで約1ヶ月かけて行ったが、その後、京コンピュータの24,000ノード(192,000 cores)でヘマグルチニン3量体(約2万4千原子系)のFMO-RI-MP2/6-31G(d)(RI-MP2:resolution of identity MP2)を行ったところ、計算時間は約11分であった。この結果は、数万原子系の構造最適化計算や短時間の分子動力学計算が近い将来現実的になることを示している。この例が示すように、巨大分子の電子状態計算は、高性能コンピュータを用いて短時間で終了する一方、構造モデルの作成、入力データの作成と結果の整理(可視化を含む)に多大な時間を費やさなければならない現実がある。これを効率化することが、電子状態計算による化学現象の研究を推進するために、今後ますます重要になると考えている。FMO計算については、構造モデリング、入力データ作成と計算結果のグラフ化を支援するGUIプログラム”fu”の開発を進めている(このプログラムのダウンロード先などの情報はMateriApps(http://ma.cms-initiative.jp/ja)を参照)。
経歴
- 1976 大阪市立大学大学院博士課程修了
- 1979 分子科学研究所助手
- 1981 大阪市立大学理学部助手
- 1989 分子科学研究所電子計算機センター助教授
- 1993 大阪府立大学総合科学部教授
- 2001 産業技術総合研究所計算科学部門総括研究員
- 2007 京都大学薬学研究科教授
- 2011 神戸大学システム情報学研究科特命教授
所属学会
- 日本化学会
- 日本薬学会
主要論文・著書
- K.Kitaura, K.Morokuma, New energy decomposition scheme for molecular interactions within the Hartree-Fock approximation, Intern.J.Quantum Chem.,10,325-340(1976).
- K.Kitaura, E.Ikeo, T.Asada, T.Nakano, M.Uebayasi,Fragment molecular orbital method: An approximate computational method for large molecules, Chem.Phys.Lett.,313,701-706(1999).
- D.G.Fedorov,N.Asada, I.Nakanishi, K.Kitaura,The Use of Many-Body Expansions and Geometry Optimizations in Fragment-Based Methods, Acc. Chem. Res.,47,2846-2856(2014).